Dear Eclipse Community,
I know there is a tremendous amount of information online, and StackOverflow full of Eclipse-RCP questions already. But the downside of that is that there is a lot of noise. Would it be an idea to set up a StackExchange dedicated to development of Eclipse RCP-based applications?
If you like the idea, please contribute to the Definition process. And if you don't, you're more than welcome to express that too :)
Thursday, June 17, 2010
Monday, May 10, 2010
How to use GitHub for [CDK|Bioclipse] code review
Triggered by posts in the past three days, I though about writing up a short tutorial on how to perform code review for existing code on GitHub. Therefore, this applied to CDK and Bioclipse source code, many but will work for any project hosted in GitHub. Even if it is not, you could consider putting up a copy there yourself. This example will demonstrate the procedure on CDK functionality in Bioclipse in the bioclipse.cheminformatics repository.
Click on the images to get a higher resolution version.
Step 1: find the class you want to review
Use the GitHub web interface to browse your way towards the source code of the class you want to review. For example, the SmartsMatchingHelper.java:
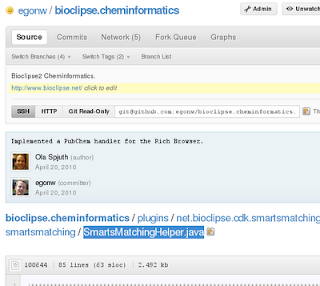
Step 2: identify something you like to comment on
Next step is to perform some code reviewing. For example, we might want to ask something about how parseProperty() works:
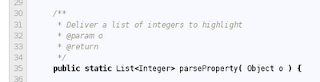 Now, this page on GitHub does not provide the means to leave comments; instead, you comment on commits.
Now, this page on GitHub does not provide the means to leave comments; instead, you comment on commits.
Step 3: find the last commit that touched the line you like to comment on
Git has a blame option (also called annotate) which will show you for each line who last changed that line. The GitHub web page makes this functionality available with the 'blame' link just above the first line of the source code:
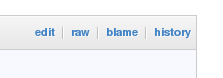 This link will lead us to a page with a new column on the left side showing commit hashes, name of the commit author, and the first few characters of the commit message. For example, the web page bits relevant to code we want to comment on, looks like:
This link will lead us to a page with a new column on the left side showing commit hashes, name of the commit author, and the first few characters of the commit message. For example, the web page bits relevant to code we want to comment on, looks like:
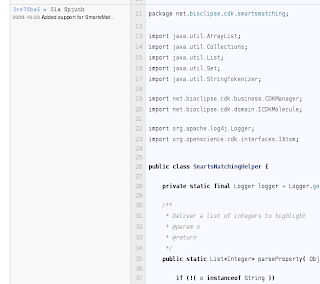 This shows us that commit 3ce78ba5 is the one we are interested in:
This shows us that commit 3ce78ba5 is the one we are interested in:

Step 4: Look up the line again and add a comment
In the web page with the appropriate commit looked in the previous step, you scroll down to the line you want to comment on. If you hover over that line, a blue comment bubble will show up on the left side:
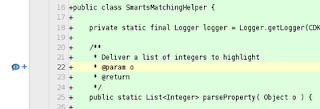 Clicking that blue comment icon, you get a dialog where you can enter your comment:
Clicking that blue comment icon, you get a dialog where you can enter your comment:
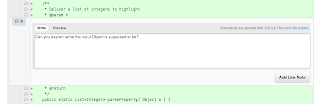 The 'Add Line Note' button confirms and saves your comment:
The 'Add Line Note' button confirms and saves your comment:
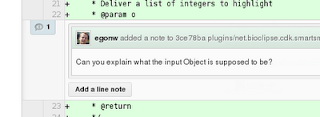
Step 5: inform the commiter about your review
The next step would be to inform the commit author. GitHub actually helps here, and should send a message, like this one:
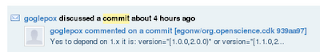 But it would certainly not hurt of you filed a bug report or sent an email.
But it would certainly not hurt of you filed a bug report or sent an email.
Now, I should only convert this into a screencast...
Click on the images to get a higher resolution version.
Step 1: find the class you want to review
Use the GitHub web interface to browse your way towards the source code of the class you want to review. For example, the SmartsMatchingHelper.java:

Step 2: identify something you like to comment on
Next step is to perform some code reviewing. For example, we might want to ask something about how parseProperty() works:
 Now, this page on GitHub does not provide the means to leave comments; instead, you comment on commits.
Now, this page on GitHub does not provide the means to leave comments; instead, you comment on commits.Step 3: find the last commit that touched the line you like to comment on
Git has a blame option (also called annotate) which will show you for each line who last changed that line. The GitHub web page makes this functionality available with the 'blame' link just above the first line of the source code:
 This link will lead us to a page with a new column on the left side showing commit hashes, name of the commit author, and the first few characters of the commit message. For example, the web page bits relevant to code we want to comment on, looks like:
This link will lead us to a page with a new column on the left side showing commit hashes, name of the commit author, and the first few characters of the commit message. For example, the web page bits relevant to code we want to comment on, looks like: This shows us that commit 3ce78ba5 is the one we are interested in:
This shows us that commit 3ce78ba5 is the one we are interested in:
Step 4: Look up the line again and add a comment
In the web page with the appropriate commit looked in the previous step, you scroll down to the line you want to comment on. If you hover over that line, a blue comment bubble will show up on the left side:
 Clicking that blue comment icon, you get a dialog where you can enter your comment:
Clicking that blue comment icon, you get a dialog where you can enter your comment: The 'Add Line Note' button confirms and saves your comment:
The 'Add Line Note' button confirms and saves your comment:
Step 5: inform the commiter about your review
The next step would be to inform the commit author. GitHub actually helps here, and should send a message, like this one:
 But it would certainly not hurt of you filed a bug report or sent an email.
But it would certainly not hurt of you filed a bug report or sent an email.Now, I should only convert this into a screencast...
Thursday, March 25, 2010
A GSoC project idea around the Resource Description Framework
I just added an entry to the Google Summer of Code 2010 Ideas wiki page:
- Resource Description Framework (RDF) is ~10 year old W3C standard. Uptake is taking off now, and it would be nice to see a Eclipse project like the Web Tools Package to provide basic RDF related functionality. This would include bundles for RDF libraries (Jena or OpenSesame) and editors for Notation3 and RDF/XML, and perhaps support for a catalog of common ontologies (RDF, RDFS, OWL, DublinCore, FOAF, ...). It could also include a Zest-based RDF graph viewer, SPARQL query editor, etc. There is existing code, for example, developed by the Bioclipse team using Jena, or the older Tripclipse. There also exist commercial offerings stressing the relevance of the RDF platform, such as Semantic Toolkit and the popular TopBraid.
Tuesday, March 16, 2010
IFile.getContentDescription() returns null on files from the workbench. Advice?
When Bioclipse reads filed its workspace, it used IFile.getContentDescription() in version 2.0 and 2.2. However, I now note that unit tests that use this method fail where they used to work in earlier versions. Instead of returning something, I get a null. An example unit test looks like:
The JavaDoc lists this methods as more efficient alternative:
As it used to work, I am considering the option it is a bug. But at the same time, maybe best practices have change? Should I keep using this method, explore the cause, perhaps file a bug report, or start using getDescriptionFor(getContents(), getName(), IContentDescription.ALL)?
propane = cdk.loadMolecule(path); Assert.assertNotNull(propane.getResource()); Assert.assertTrue(propane.getResource() instanceof IFile); IFile resource = (IFile)propane.getResource(); Assert.assertNotNull(resource.getContentDescription()); IContentType type = resource.getContentDescription().getContentType(); Assert.assertNotNull(type); IChemFormat format = cdk.determineFormat(type); Assert.assertNotNull(format); Assert.assertEquals(MDLV2000Format.getInstance(), format);This test uses the getContentDescription() to get a content description and converts it to a CDK library specific format type.
The JavaDoc lists this methods as more efficient alternative:
- Calling this method produces a similar effect as calling getDescriptionFor(getContents(), getName(), IContentDescription.ALL) on IContentTypeManager, but provides better opportunities for improved performance.
As it used to work, I am considering the option it is a bug. But at the same time, maybe best practices have change? Should I keep using this method, explore the cause, perhaps file a bug report, or start using getDescriptionFor(getContents(), getName(), IContentDescription.ALL)?
Monday, March 15, 2010
RDF-powered QSAR wizard: SPARQL end points providing wizard content
As you know from my blog, one of the things I am working on is to push RDF functionality in Bioclipse, as I believe it to be the missing link between molecular chemometrics and literature, databases, and other non-numerical information sources.
As part of the submission for the SWAT4LS special issue in the new Journal of Biomedical Semantics, Ola hacked up a cool wizard that sets up a new QSAR Project by downloading data directly from our RDF node for the chEMBL data using SPARQL. The paper is based on the SWAT4LS talk I gave, and the proceedings paper that recently appeared. But with more cool stuff, such as this cool RDF graph browser that allows you to open up molecules from the RDF graph in a JChemPaint editor.
Well, this really nice New QSAR Project wizard was cool enough to trigger a I-want-more reaction, so I just had to hack it up with some additional SPARQL functionality. So, the next version does not only use RDF and SPARQL to aggregate the QSAR data set, it also uses SPARQL to make the wizard interactive. While the user is typing a target ID, the wizard will check the SPARQL end point in the background and download the target's type, title and organism, as well as update the list of activities the user can select depending on what the chEMBL database has for that target:
 The actual code base is pretty small, and that's what happens when you mash up the right technologies :)
The actual code base is pretty small, and that's what happens when you mash up the right technologies :)
As part of the submission for the SWAT4LS special issue in the new Journal of Biomedical Semantics, Ola hacked up a cool wizard that sets up a new QSAR Project by downloading data directly from our RDF node for the chEMBL data using SPARQL. The paper is based on the SWAT4LS talk I gave, and the proceedings paper that recently appeared. But with more cool stuff, such as this cool RDF graph browser that allows you to open up molecules from the RDF graph in a JChemPaint editor.
Well, this really nice New QSAR Project wizard was cool enough to trigger a I-want-more reaction, so I just had to hack it up with some additional SPARQL functionality. So, the next version does not only use RDF and SPARQL to aggregate the QSAR data set, it also uses SPARQL to make the wizard interactive. While the user is typing a target ID, the wizard will check the SPARQL end point in the background and download the target's type, title and organism, as well as update the list of activities the user can select depending on what the chEMBL database has for that target:

Thursday, March 4, 2010
RDF, Jena, Bioclipse, Eclipse, Zest #2: icons and an extension point
Jonathan worked this week on new features for the Bioclipse RDF editor (see these two earlier items). This version still does not edit, but only display using Zest. Jonathan created for me an extension point so that anyone can make the editor aware of domain objects, by simply registering the extension implementation along with the rdf:Class URI of the rdf:type of an object. This fixes the problem of having to hardcode dependencies of the RDF editor on all the domain code, as was the case earlier.
For example, the cheminformatics IMolecule object is now linked to the rdf:type <http://www.bioclipse.net/structuredb/#Molecule>:
 For example, double clicking the ron:mol2 node, would open up a JChemPaint editor.
For example, double clicking the ron:mol2 node, would open up a JChemPaint editor.
For example, the cheminformatics IMolecule object is now linked to the rdf:type <http://www.bioclipse.net/structuredb/#Molecule>:
<extension point="net.bioclipse.rdf.rdf2bioobjectfactory">
<Factory
instance="net.bioclipse.rdf.ui.RDFToCDKMoleculeFactory"
uri="http://www.bioclipse.net/structuredb/#Molecule" >
</Factory>
</extension>
The API for this factory looks like:public IBioObject rdfToBioObject( Model model, Resource res ); public ImageDescriptor getImageDescriptor();This is very much tied into the Jena data model, so not entirely clean, but has to do for now. The first method converts RDF content into a Bioclipse IBioObject, such as an IMolecule (see this list of currently supported objects). The second method returns an icon, which makes the editor more visually pleasing, and provides a nice way to see when you can double click the RDF node to have it open in an domain specific editor:

Sunday, February 7, 2010
RDF, Jena, Bioclipse, Eclipse, Zest: Mashups
Quite a while a go, I blogged about Zest in Bioclipse showing a bit of ONS Solubility data. I could not follow up on that until now, as I had yet to do a lot of RDF work in Bioclipse, so the screenshot back then was kind of a mockup.
Things are different now, and the Bioclipse-RDF functionality (using Jena) is released in Bioclipse 2.2 (see Semantic Web features in Bioclipse 2.2), and I got around to writing the graphical goodies for the following papers. Not submitted yet, but here's the screenshot showing a N3 file opened with a Zest-powered editor (read-only) and a plain text editor:

Things are different now, and the Bioclipse-RDF functionality (using Jena) is released in Bioclipse 2.2 (see Semantic Web features in Bioclipse 2.2), and I got around to writing the graphical goodies for the following papers. Not submitted yet, but here's the screenshot showing a N3 file opened with a Zest-powered editor (read-only) and a plain text editor:

Wednesday, February 3, 2010
Semantic Web features in Bioclipse 2.2
Ola is releasing Bioclipse 2.2.0 today, and asked me to show case the semantic web functionality in Bioclipse. I realized that I do not have a nice page showing the semantic web overview. But I did blog a lot about RDF functionality, so here's a list of pointers:

One thing I have not blogged about yet (I think), is that the Bioclipse RDF manager also understands RDFa now. Well, sort of... it relies on a webservice, but this is what the script looks like:
- Bioclipse Manager for MyExperiment.org
- Bioclipse, RDF and defeasible reasoning (see also Samuel's blog)
- Bioclipse and SPARQL end points #2: MyExperiment
- Bioclipse and SPARQL end points
- Solubility Data in Bioclipse #2: handling RDF
- Solubility Data in Bioclipse #3: Finding ChEBI IDs
- Solubility Data in Bioclipse #4: Finding ChEBI IDs (Again, but better)
- /me is having Bioclipse/XMPP/RDF fun
One thing I have not blogged about yet (I think), is that the Bioclipse RDF manager also understands RDFa now. Well, sort of... it relies on a webservice, but this is what the script looks like:
model = rdf.createStore() rdf.importRDFa(model, "http://egonw.github.com/") rdf.saveRDFN3(model, "/Virtual/egonw.n3")With support of SPARQL end points, and reading RDF from web resources directly (RDF/XML, N3, RDFa), Bioclipse is ready for the chemical semantic web.
Monday, February 1, 2010
Validating MDL SD files and Symyx molfiles with the CDK
Bioclipse 2.0 introduced a new, powerful molecular table support, and we have been eager to test that on large SD files. A recent ChEBI SD file failed to open, and eyes were immediately at the CDK, which is the cheminformatics library used in Bioclipse.
After careful investigations, it turned out that the ChEBI file contained a few entries which were not MDL molfiles, but queries for the ISISBase system. Those cannot be read by the CDK MDLV2000Reader. However, it crashed on it, instead of failing more savely. That's not nice, and fixed. But, the problem is rather recurrent, and the reason why I like CML so much: invalid input. CML, based on XML, has several general validation approaches that give in-depth error messages of what is wrong with the file.
So, I asked on the BOx what the Open Source cheminformatics community had to offer for this. Turns out that several tools find problems in the files, but none could report where the error occurred.
Validation
Now, some time ago, I played with two reading modes, RELAXED and STRICT, as faulty files is core cheminformatics material, and the software is blamed if the QSAR model resulting from it is not good (seriously). Anyways, a small API change in the CDK would make a validating MDLV2000Reader quite a step closer, but I had not followed up on it until last Friday where I patch I was reviewing caused 6 new unit test fails. The new fails were caused by a assumption which turned out the be false in the test files used in those 6 unit tests.
The MDL (or Symyx) molfile specifications (not an Open Specification) defines an atom block line as:
Now, personally, I rather send the file back to the user with a proper error report and show them what is wrong with the file. Or better, provide them with a MDL V2000 text editor (e.g. in Bioclipse) which would graphically highlight errors, as many of us are used to with Eclipse:

CDK Patch
So, I am hacking up a patch for CDK master to allow error reporting by IChemObjectReaders. The initial version of the API update and use in the MDLV2000Reader are available as Gist 290659. They are not final yet, as I realized when making the above screenshot, that merely int col is not enough, and that I actually need the startCol and endCol positions instead. Also, there are only an error level at this moment, and no warning level as in the screenshot.
That said, I created a jar (ant dist-large) and saved it as mdlCheck.jar, and wrote a bit of Groovy:
which defines a class implementing the new IChemObjectReaderErrorHandler and then reads a MDL molfile. And the output looks like it fulfills my needs:
Or, another common found issue (using D and T as element symbols):
Enough for now... dinner time.
After careful investigations, it turned out that the ChEBI file contained a few entries which were not MDL molfiles, but queries for the ISISBase system. Those cannot be read by the CDK MDLV2000Reader. However, it crashed on it, instead of failing more savely. That's not nice, and fixed. But, the problem is rather recurrent, and the reason why I like CML so much: invalid input. CML, based on XML, has several general validation approaches that give in-depth error messages of what is wrong with the file.
So, I asked on the BOx what the Open Source cheminformatics community had to offer for this. Turns out that several tools find problems in the files, but none could report where the error occurred.
Validation
Now, some time ago, I played with two reading modes, RELAXED and STRICT, as faulty files is core cheminformatics material, and the software is blamed if the QSAR model resulting from it is not good (seriously). Anyways, a small API change in the CDK would make a validating MDLV2000Reader quite a step closer, but I had not followed up on it until last Friday where I patch I was reviewing caused 6 new unit test fails. The new fails were caused by a assumption which turned out the be false in the test files used in those 6 unit tests.
The MDL (or Symyx) molfile specifications (not an Open Specification) defines an atom block line as:
xxxxx.xxxxyyyyy.yyyyzzzzz.zzzz aaaddcccssshhhbbbvvvHHHrrriiimmmnnneeebut does not specify which fields are optional. And indeed, many tools around save MDL molfiles with one or more fields missing, leading to shorter than expected line lengths. And, as you might have expected, the failing unit tests had files with lines missing the field introduced by the patch, causing Exceptions being thrown around. I have yet to make up my mind of the lack of those fields is a problem in the file, or allowed by the format. In either case, the information from that field is not available, and the reader could safely ignore the missing information. Per user demand.
Now, personally, I rather send the file back to the user with a proper error report and show them what is wrong with the file. Or better, provide them with a MDL V2000 text editor (e.g. in Bioclipse) which would graphically highlight errors, as many of us are used to with Eclipse:

CDK Patch
So, I am hacking up a patch for CDK master to allow error reporting by IChemObjectReaders. The initial version of the API update and use in the MDLV2000Reader are available as Gist 290659. They are not final yet, as I realized when making the above screenshot, that merely int col is not enough, and that I actually need the startCol and endCol positions instead. Also, there are only an error level at this moment, and no warning level as in the screenshot.
That said, I created a jar (ant dist-large) and saved it as mdlCheck.jar, and wrote a bit of Groovy:
which defines a class implementing the new IChemObjectReaderErrorHandler and then reads a MDL molfile. And the output looks like it fulfills my needs:
$ CLASSPATH=mdlCheck.jar groovy mdlCheck.groovy src/test/data/mdl/test6.sdf location: 5, 35: Could not parse mass difference field. -> For input string: "" location: 6, 35: Could not parse mass difference field. -> For input string: ""Note to myself, that atom block does not like like a MDL molfile atom block at all! Every second line outputs the Exception passed to the error handler. I have to say, those messages are rather cryptic, but resulting from a NumberFormatException, if not mistaken.
Or, another common found issue (using D and T as element symbols):
$ CLASSPATH=mdlCheck.jar groovy mdlCheck.groovy src/test/data/mdl/hisotopes.mol location: 6, 32: Invalid element type. Must be an existing element, or one in: A, Q, L, LP, *. location: 7, 32: Invalid element type. Must be an existing element, or one in: A, Q, L, LP, *.
Enough for now... dinner time.
Subscribe to:
Posts (Atom)